扩展共轭双键怎么判断
阅读文章前辛苦您点下“关注”,方便讨论和分享,为了回馈您的支持,我将每日更新优质内容。

文 |史记新说
编辑 | 史记新说
简介
这种清洁能源生产是21世世纪.由于使用化石燃料,全球能源吸引力日益增加以及对全球变暖的显著关注鼓励了新技术和能源载体的发展。
在各种能量载体系统中,H2是环保和绿色环境的知名和最清洁的能源载体。已经报道了许多制氢/储存系统,即金属/化学氢化物,无机和有机纳米管和金属有机框架。
在过去的几十年中,众所周知,非无辜配体充当有机金属催化剂并在生物框架中起作用。在非无辜配体中报告的异常异常歧化在双电子过程中产生稳定的氧化剂和还原剂。
对于还原或氧化,氧化还原活性配体具有更主动可及的水平。相比之下,早期关于功能有机分子过渡金属配合物中质子,电子或氢化物位移的报告使我们能够揭示由质子/电子(H / e)支持的化学反应的分子见解+−)转移机制。
这些分子系统可能提供扩展新型多步H / e的选择+–运动过程以及生成H的结果2-存储/-进化材料.Peng等人描述了邻苯并醌二胺(bqdi)和半邻苯并醌二胺(s-bqdi)基铁(II)、钴(II)和钴(III)配合物的键型,[Fe第二(拜迪)3](PF6)2, [Co第二(S-BQDI)2]和公司第三Cl(s-bqdi)2。
如方案1所示,opda配体与过渡金属离子配位产生相应的氧化产物,
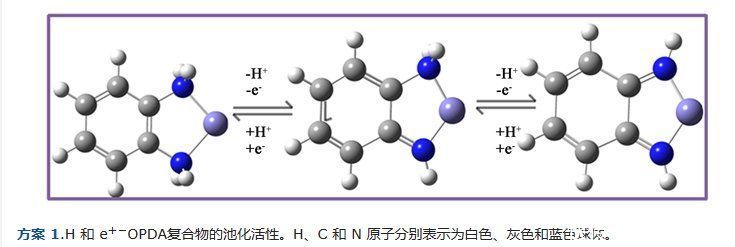
邻二亚氨基苯醌(SBQDI)和邻二亚氨基苯醌(BQDI)通过简单的氧化。游离化合物总共可以捐献两个质子(2H+)和两个电子(2e−)的各种组合。
氧化形式bqdi与过渡金属Fe(II)配位并形成tris [Fe(bqdi)3]2+复杂。同一组研究人员还报告了混合bpdi-opda钌金属配合物的合成和晶体学研究,[Ru第二(奥普达)2(BQDI)][PF6]2。
他们还报告了使用金属辅助配体氧化[Ru第二(奥普达)(拜迪)2][PF6]2to [Ru(bqdi)3]2+.最近,Matsumoto及其同事报告了非贵金属辅助H2由氧化还原活性和非无害配体邻苯二胺(OPDA)及其Fe(II)配合物生产。
最近,他们合成了一种低自旋(ls)铁(II)-bqdi配合物ls-[Fe第二(拜迪)3][PF6]2,显示室温下THF溶剂中的光化学析氢(PHE)活性。
这种类型的金属配合物可能会在需要多个质子和电子转移的化学过程中揭示与能量相关的反应的功能,如水分解,制氢和固氮。
然而,尚未报道与bqdi配体形成tris配合物的第一行二价转变系列的系统研究。因此,基于这项简短的调查,本研究的目的是研究与bqdi配体[M(bqdi) [M(bqdi)3]2+([M = Ti2+到锌2+])在B3LYP/6-311G(d,P)水平上使用密度泛函理论(DFT)。
具体来说,为了详细了解Fe-bqdi-tris复合体,[Fe(bqdi)3]2+,我们系统地描述了紫外和前沿分子轨道(FMO)。
计算细节
三(邻苯二胺)M(II)的几何优化(其中M = Ti2+到锌2+)使用密度泛函理论(DFT)和B3LYP/6-311G(d,p)理论水平在THF中进行配合物。
本模拟采用了Gaussian16仿真包和GaussView 6.0。应用于优化几何图形的可视化。当最大原子力分别小于0.00045 Hartree/Bohr和最大位移至阈值0.0018 Bohr时,考虑几何优化。
没有对 [M(bqdi) 施加对称约束3]2+.溶剂的影响由导体状极化连续溶剂化模型(CPCM)考虑。过渡金属原子采用LANL2DZ有效核心电位(ECP),其他原子采用6-311G(d,p)基集。
通过检查正常模式坐标和无负频率来发现最低能量结构。使用优化的几何形状执行时变(TD)DFT计算,以评估THF中平衡结构的垂直激发能,采用CAM-B3LYP [26] /6-311++ G(d,p)理论水平以及远程校正。使用以下公式计算了配位的金属-配体结合能。等式(1)中表示:
其中物种E复杂,E金属和E配体表示 [M(bqdi) 的能量3]2+(其中 M = Ti2+到锌2+)配位配合物,金属离子和配体邻苯并醌二胺(BQDI)。
因此,结合能,ΔE状态为每个配体。每个配位复合物和配体都单独优化。使用高斯程序中包含的NBO 3.1软件,计算了电子结构,自然键序(NBO)和全局描述符。
针对电子电荷和主要供体-受体相互作用,对优化结构进行了NBO研究。过渡金属的电荷使用不同的收费方案计算。
结构分析
tris-[M(bqdi) 的几何参数3]2+(M = Ti-Zn离子)配合物的计算和总结见表1。图1显示了三(bqdi)过渡金属(II)配合物的优化结构。每个金属配合物的优化几何形状都是由三个双齿bqdi配体组成的扭曲八面体。
bqdi配体-金属配合物的所有几何形状都与[Fe(bqdi)相同3]2+ [[14]),尽管金属-氮(M-N(bqdi))的距离略有不同(表1)。
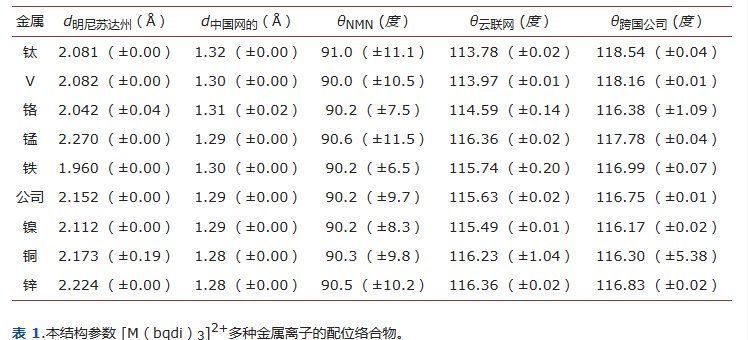
金属和氮原子的平均相距为1.960至2.270 Å。所研究配合物中的Mn-N键距离[Mn(bqdi)3]2+在 2.270 - 2.273 Å 的范围内,并且悄悄地比 [Fe(bqdi) 中的 Fe-N 距离长3]2+(1.957 - 1.958 Å)。
彭等在他的X射线几何分析中报告了Fe-N的键长[Fe(bqdi)3]2+(PF6)2复合体范围从 1.906 - 1.925 Å。
Tris bqdi配合物中Fe-N的平均键长为1.916 Å 。松本等.找到键距的 Fe-N 范围为 1.919 - 1.931 Å 的 [Fe(bqdi)3]2+复杂 .M-N在其他三邻苯并醌二胺(bqdi)配合物中的距离略高于[Fe(bqdi)3]2+, 2.076 - 2.078 Å 代表 [Ti(bqdi)3]2+, 2.075 - 2.076 Å 代表 [V(bqdi)3]2+, 2.003 - 2.085 Å 代表 [Cr(bqdi)3]2+, 2.149 - 2.191 Å 代表 [Co(bqdi)3]2+, 2.047 - 2.416 Å 为 [Cu(bqdi)3]2+和 2.219 - 2.221 Å 表示 [Zn(bqdi)3]2+复合体,分别。计算出的这些配合物的金属-氮(M-N)距离与配合物的结合强度相关。
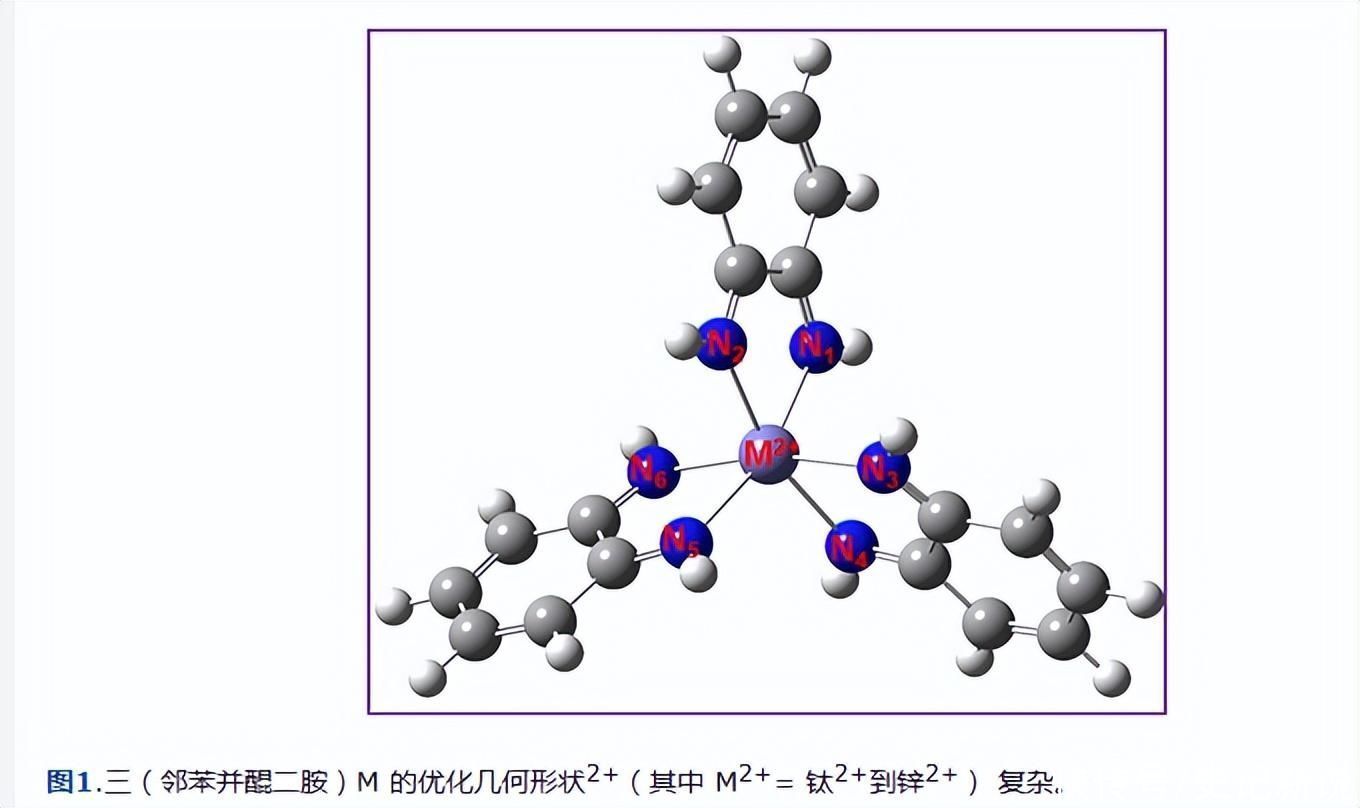
在这项工作中,计算出的N-Fe-N键角主要大于先前的X射线晶体结构81.26°[14]。此外,图1还说明了与中心铁原子的三角畸变的八面体配位几何形状。
从邻苯并醌二胺(bqdi)配体中发现的“咬合”以这样一种方式被显着和施加,即N-Fe-N角从90.0°到79.55°被压实。极小的Fe-N距离(平均1.960 Å)表明了令人难以置信的强结合,具有低自旋和bqdi配体的相关铁。
邻苯并醌二胺(bqdi)配体的几何形状是平面的。六个C=N键(平均l.300 Å)和它们的六个C=C共轭键(平均1.354 Å)很小,与它们被识别为局部双键一致。卡波夫斯基等.发现计算出的 C=C 键为 1.357 Å。
挥之不去的十二个C-C键长度(~1.445 Å)仅比环辛四烯(1.46 A)中的C-C单键略小。该复合物中bqdi配体的计算键类型与[Fe第二-(中国),(bqdi)]2− , [俄罗斯第二(拜迪)(比皮)]2+,并且比[Ni(s-bqdi)2]。
邻苯并醌二胺(bqdi)部分的键原型在上述每个复合物中也相似,并且比中性或纯邻苯并醌二胺(bqdi)配体复合物具有额外的离域双键。
结合能分析
在这项工作中,我们系统地研究了第一排二价过渡金属离子的结合能ΔE,焓(ΔH)和吉布斯自由能(ΔG)。tris 的结合能 (ΔE)、焓和吉布斯自由能 [M(bqdi)3]2+(其中M = Ti-Zn)具有不同自旋态的配合物计算并在表2中制表。
负结合能(ΔE)与类似配合物的持久性相互关联。这说明了THF溶剂中配合物的耐久性随着最高结合能的提高而加速。电子能仅用于估计上述ΔE的结合能。由于金属-lgand结合本质上是共价的,因此,对ΔE的振动,热和熵贡献是微不足道的。
由于具有不同自旋态的配合物的稳定性,计算出的结合能趋势略有不同。在这项研究中,Fe2+具有较低自旋的BQDI配体配合物比其他自旋的Fe高度稳定2+。
图2显示了金属-配体结合能与金属核电荷的双驼峰特性。由于电子相互作用的增加,总的来说,随着核电荷的增加而增加。由于Jahn-Teller失真,ΔE从V降低到Cr配合物。有利于Mn的观点下降归因于d5Mn(II)的电子配置。
由于所有d轨道的占据,从配体到金属的电荷转移不足,导致ΔE降低。Zn(II),其具有 d10电子配置也显示出效率低下。因此,对于Zn(II)来说,这是相对较少的。双驼峰特性与六水复合物观察到的特征相似。
请注意,本研究目前计算的ΔE有两个峰位于V和Cu。这两个峰的出现是由于高配体场稳定性有利于3和 d9高自旋配合物的配置。
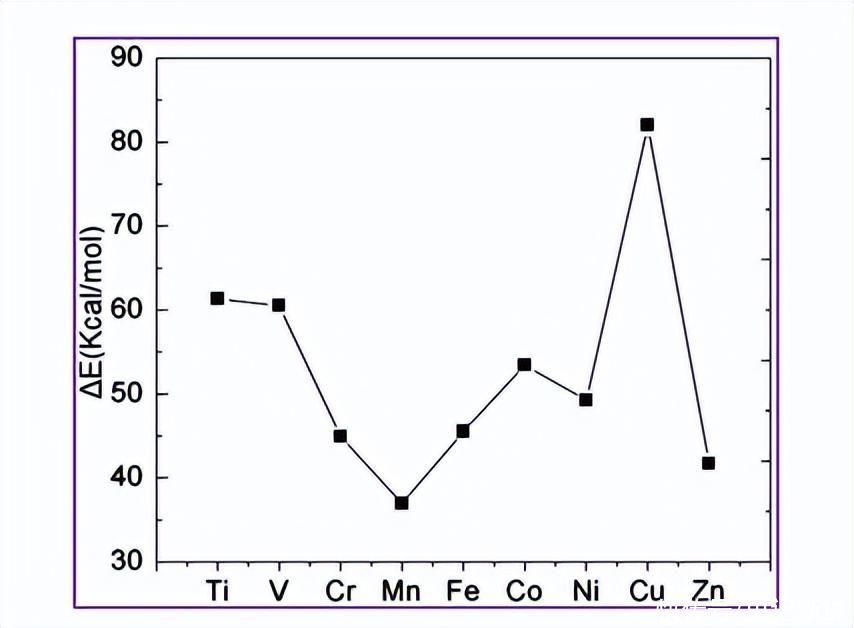
图2.计算出的过渡金属与bqdi配体配位的结合能。在不施加八面体对称性的情况下,计算每个配位复合物的每配体结合能∆E。
考虑与BQDI配体协调的其他金属离子。因此,在Fe(II)存在下检查的bqdi配体的强结合应源于其他因素。ls(低自旋)态的Fe(II)比hs态高3.28 kcal∙mol−1 (表 2)。
考虑到能量,筛选了ls >hs阵列中复加速度的持久性。通过评估结合能,得出结论,ls态的Fe(II)是形成tris [Fe(bqdi)3]2+复杂。遵循Boys-Bernardi对位(CP)[34]校正方法,它确认了基集叠加误差(BSSE)以支持tris [Fe(bqdi)3]2+仅具有低自旋状态的复杂。
修正后的BSSE能量ECPcomplex(−1148.47 au)与未校正的能量进行称重Ecomplex(−1730.06 AU)。比较题外话的定义
计算出的BSSE能量在0.012%以内,这是计算误差的范围。这里只包括电子能来计算结合能。由于金属配体的结合本质上是共价的,振动的,热贡献和熵贡献被逐出为分钟。
此外,计算出的结合焓(ΔHbind)和吉布斯自由能(ΔGbind)的配位表明,配体bqdi和Fe(II)离子在低自旋时的相互作用高于高自旋和中间自旋。
注意到Fe(II)具有相同配体bqdi低自旋配合物的配合物比高自旋态更稳定。除金属离子外,ZPE、热能、焓和吉布斯自由能校正后的ΔE均在室内不准确结合能的6%。
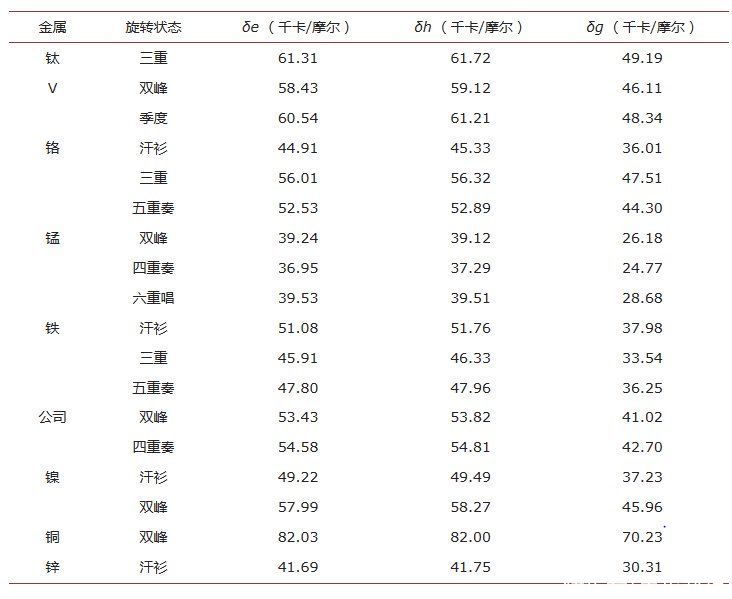
表 2.目前计算的金属-配体结合能、焓和吉布斯自由能[M第二(拜迪)3]2+协调的复合体。
结论
本工作从理论上描述了三(邻苯并醌二胺)配体的第一排二价过渡金属配合物的比较结构性质。采用DFT B3LYP/6-311G(d,P)理论水平计算了结合强度、带隙能量、电荷、NBO相互作用强度和全局反应性描述子分析。
金属-配体结合能是金属和bqdi配体之间协调的主要分子基础之一。本结果表明,激活第一行二价过渡金属离子负载的有机部分的最可能方法。
本结果还表明,具有低自旋态的Fe(II)的结合强度不足以与配体bqdi形成稳定的配合物。
计算值与以往实验结果吻合较好。该结果将为未来研究此类配体系统的配位建模动力学提供参考。
","gnid":"960ff70df9874eea3","img_data":[{"flag":2,"img":[{"desc":"","height":"121","title":"","url":"https://p0.ssl.img.360kuai.com/t0105c12b150cb3e03d.jpg","width":"767"},{"desc":"","height":"336","title":"","url":"https://p0.ssl.img.360kuai.com/t01893efb1d831327d3.jpg","width":"900"},{"desc":"","height":"247","title":"","url":"https://p0.ssl.img.360kuai.com/t01ebc1479f11cc9b24.jpg","width":"735"},{"desc":"","height":"500","title":"","url":"https://p0.ssl.img.360kuai.com/t01d6887e20e2b3afe2.jpg","width":"1731"},{"desc":"","height":"283","title":"","url":"https://p0.ssl.img.360kuai.com/t01e017de22a85a78f5.jpg","width":"395"},{"desc":"","height":"500","title":"","url":"https://p0.ssl.img.360kuai.com/t01672d3a0245488b80.jpg","width":"1072"},{"desc":"","height":"340","title":"","url":"https://p0.ssl.img.360kuai.com/t01dbeeb6b2e9c79a22.jpg","width":"748"},{"desc":"","height":"808","title":"","url":"https://p0.ssl.img.360kuai.com/t01719716d3ce1b12b3.jpg","width":"1360"},{"desc":"","height":"324","s_url":"https://p0.ssl.img.360kuai.com/t018b1750a52020f5a7_1.gif","title":"","url":"https://p0.ssl.img.360kuai.com/t018b1750a52020f5a7.gif","width":"432"},{"desc":"","height":"494","title":"","url":"https://p0.ssl.img.360kuai.com/t019bd17c482515b60c.jpg","width":"1280"},{"desc":"","height":"628","title":"","url":"https://p0.ssl.img.360kuai.com/t014300f7a82229b89d.jpg","width":"854"},{"desc":"","height":"599","title":"","url":"https://p0.ssl.img.360kuai.com/t010dc120aab555d5a3.jpg","width":"732"}]}],"original":0,"pat":"art_src_3,fts0,sts0","powerby":"hbase","pub_time":1689666120000,"pure":"","rawurl":"http://zm.news.so.com/513a2910d11214b68b6f6e5b19c50d24","redirect":0,"rptid":"b3238de74253a22a","rss_ext":[],"s":"t","src":"史记新说","tag":[],"title":"邻苯并醌二胺-第一排二价过渡金属配合物的理论研究
闵毛江2181紫外如何判断是否有共轭体系存在
凤削舒13993455167 ______ 具有共轭双键的化合物,相间的π键与π键相互作用(π-π共轭效应),生成大π键.由于大π键各能级间的距离较近电子容易激发,所以吸收峰的波长就增加,生色作用大为...
闵毛江2181在红外光谱中如何根据分子结构式确定判断环内双键、环外双键延伸双键和共轭体系上的取代烷基希望有图和例何为无环二烯烃、异环二烯烃、同环二烯烃?... -
凤削舒13993455167 ______[答案] 找本兰氏化学手册看看有没有 如果找有机化学书 有机化学书我有 可是发图片太麻烦了
闵毛江2181氨基酸中什么是共轭双键 -
凤削舒13993455167 ______ 共轭双键是有机化合物分子结构中由一个单键隔开的两个双键,氨基酸没有共轭双键
闵毛江2181化学:共轭指什么? -
凤削舒13993455167 ______ 正常共轭效应 又称 - 共轭.是指两个以上双键(或三键)以单键相联结时所发生的 电子的离位作用.英戈尔德,C.K.称这种效应为仲介效应,并且认为,共轭体系中这种电子的位移是由有关各原子的电负性和 p 轨道的大小(或主量子数)决定...
闵毛江2181苯环上有共轭双键吗? -
凤削舒13993455167 ______ 苯环的结构是由三个极限共振式组成,每个极限共振式都有共轭双键,所以苯环也有共轭双键.大π键只是一个理论,苯环的结构是由不同理论支撑的,极限共振理论就是其一. 共轭双键就是使两个隔一个单键的双键形成类似于单键和双键之间...
闵毛江2181什么是共轭? -
凤削舒13993455167 ______ 共轭在数学、物理、化学、地理等学科中都有出现. 本意:两头牛背上的架子称为轭,轭使两头牛同步行走.共轭即为按一定的规律相配的一对.通俗点说就是孪生.在数学中有共轭复数、共轭根式、共轭双曲线、共轭矩阵等. 共轭方向法在...
闵毛江2181苯环里的π键为什么稳定? -
凤削舒13993455167 ______[答案] 共轭双键有机化合物分子结构中由一个单键隔开的两个双键.以C=C-C=C表示这类化合物很容易聚合,并能发生特殊的1,4加成反应.共轭双键是以C=C-C=C为基本单位,随着共轭度的增加,其紫外特性:最大吸收波长红移;如有荧光,...
闵毛江2181苯甲酸对位连有基团时,若存在共轭效应,如何判断其是吸电子共轭效应还是给电子共轭效应? -
凤削舒13993455167 ______ 苯甲酸中原本就存在p-π共轭效应,苯环上大π键与羧基中碳氧双键中氧原子的空轨道p共轭 吸电子共轭效应与推电子诱导效应的强弱一般情况下是共轭效应强一些,不过我还没有见过二者强弱比较的权威资料
闵毛江2181以下哪个亲电加成反应活性最大?为什么?如何比较? -
凤削舒13993455167 ______ 乙烯>丙烯>氯乙烯>2—甲基丙烯. 因为亲电试剂在进衫数攻双键时,电子云密度越高,越容易进行,卤素是吸电子原子,而且原子半径越小,吸电子能力越强,双键碳上连得卤素原子越多,电子云密度越低,反应活性就越低. 亲电加成反应的...